Author: Silvia Marinova, PhD student in the Genomic Stability Laboratory at BAS
Thalassemia is a rare genetic inherited blood disease in which the body does not produce enough of the hemoglobin protein. It is an essential part of red blood cells and is responsible for the transfer of oxygen between individual tissues in the body. When not producing a sufficient amount of it, this leads to the malfunctioning of red blood cells and shortens their lifes.
As a result, a smaller proportion of them circulate in the bloodstream, and oxygen that reaches the cells is not enough. This leads to anemia, a feeling of fatigue and other symptoms, which we will consider in more detail in the article. We will also pay attention to the types of thalassemia, how it is diagnosed and what treatment there is.
Types of thalassemia
Thalassemia is most common in people from Mediterranean, African and South-Eastern Asian populations. However, due to increased migration, it is already found in other areas [ref.1].
When talking about different types of thalassaemia, we may consider two main things. If it is the part of hemoglobin that is affected, then two main types are recognized - alpha or beta thalassemia [ref.2]. They, in turn, can be divided into subtypes depending on the severity of the disease - thalassemia minor, intermediate or major.
Hemoglobin is an iron-containing protein that carries oxygen into our body. Its main form is composed of the so-called alpha and beta globlin chains and contains two of them. If one of the chains is produced in a smaller amount or is not produced at all, then thalassemia occurs. Depending on which of the two is affected, we distinguish the alpha and beta form of the disease.
How is thalassaemia inherited
Thalassemia is inherited from parents to children, and because of this it is said that it is an inherited disease. In general, inheritance is autosomal recessive, which means that we need to inherit a defective allele of the gene from both parents in order to develop the disease. If, however, we inherit such a gene from only one parent, then we are carriers of the disease and more often symptomatology does not develop.

The HBB gene [ref.3] is responsible for the synthesis of beta globrin chains. When there is an error in it, our cells receive incorrect instructions and this leads either to their reduced amount or to the lack of their synthesis.
The gene is represented in two copies (allels) in our genome - one from each parent, and since the disease is inherited autosomally recessively, upon receiving two mutant copies, then a disease with severe symptoms develops - beta thalassemia intermedia or major.
If we are carriers with only one damaged gene, then in most cases there is no symptomatology, but it is possible in some cases to develop the mild "minor" form [ref.4].
Read more about heredity in the article
Inheritance in alpha thalassaemia is a little more complicated, since alpha globine chains need two genes HBA1 [ref.5] and HBA2 [ref.6]. For each gene, one copy is inherited from the mother and the other from the father. If at least one copy of the gene is divided (missing) in both parents, then there is a risk that their children will develop alpha thalassaemia. The severity of the disease depends on how many copies are divided and in what combination.
- One deleted copy - this is the mildest form of alpha thalassemia, and more often individuals with this type of mutation are only carriers. In rare cases, non-severe microcytosis [ref.7] develops, which is characterized by smaller than normal red blood cells (erythrocytes).
- Two deleted copies - when two copies are missing, this also leads to a mild state of alpha thalassemia minor.
- Three deleted copies - then an average disease develops in severity, which is known by the name HbH thalassemia. The result of it is an excess of beta globin chains and their aggregation in the form of abnormal aggregates in red blood cells.
- Four deleted copies - this is the most severe form in which alpha chains are not produced at all. The disease is called Hb Barth syndrome and leads to severe changes during fetal development. In most cases, this causes premature birth, and even if not - the baby rarely survives more than a few months after birth.
What are the symptoms of thalassemia
The clinical picture of the disease depends on the type of thalassemia, as well as its severity.
Symptoms may include:
- general fatigue
- weakness
- pale or yellowish skin
- enlarged and deformed bones, especially facial
- dark urine
- oxygen deficiency (hypoxia)
- delayed growth and development, etc.
Often the symptoms are visible at birth or develop in the first and second year of the baby's life. In milder forms, symptoms may also appear in later childhood or adolescence.
Diagnosis and prevention of thalassemia
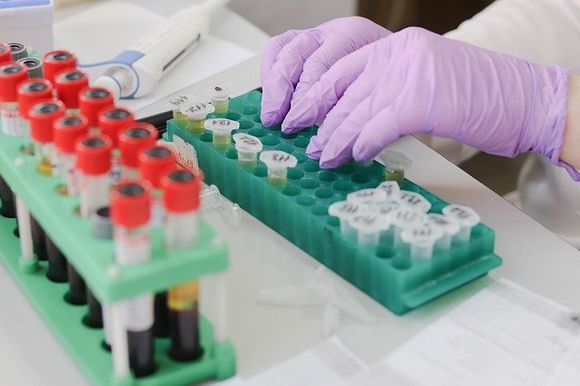
Diagnosis usually begins with a blood test. The doctor will monitor indicators such as amount of hemoglobin, erythrocyte size, bilirubin level, iron in the blood and others. To confirm the diagnosis, the so-called hemoglobin electrophoresis is performed, which can indicate the ratio between the different globin chains.
There are also genetic tests that can confirm the type of thalassemia. If it is known that there is a family history of the disease, then such a test can be done to future parents who plan to have a child in order to assess the risk to offspring.
Treatment of thalassemia
Methods of treatment are again directly dependent on the severity of the condition. People who suffer from this disease should be treated by doctor whose specialty is associated with blood diseases - a hematologist.
In severe forms of the disease, patients usually need frequent blood transfusions and/or iron-chelating therapy [ref.8] to bind excess iron in the blood and thus prevent serious organ damage. People suffering from milder forms often do not need specific treatment.
It should be noted that it is extremely important for all patients with thalassemia to make "healthy" choices, because for them this means dealing with the disease and improving their quality of life. It is advisable to observe a specific diet in which to avoid iron-rich foods, to do exercises tailored to the severity of the disease and to make all mandatory and recommended immunizations, since people with thalassemia have a higher risk of developing certain infections.
Read more:
How genes determine resemblance to our parents
How nucleic acids DNA and RNA orchestrate life
Does it make sense to take a genetic test?