Author: Rositsa Tashkova, Master of Molecular Biology and Microbiology
Thalassemia and sickle cell anemia are genetic diseases of the blood that are inherited and therefore cannot be cured, but nevertheless there are ways to maintain the better general health of those suffering from them.
In both diseases, mutations are in the genes for hemoglobin - the protein that binds and carries oxygen using erythrocytes (red blood cells). For very severe cases of these diseases, hope is given by the new CRISPR-Cas9 technology, as well as stem cell therapy.
Read more in the articles and .
- CRISPR-Cas9 technology - is it ethical and can it be used as a treatment
- Stem cell transplantation as cancer treatment
What is Sickle Cell Disease
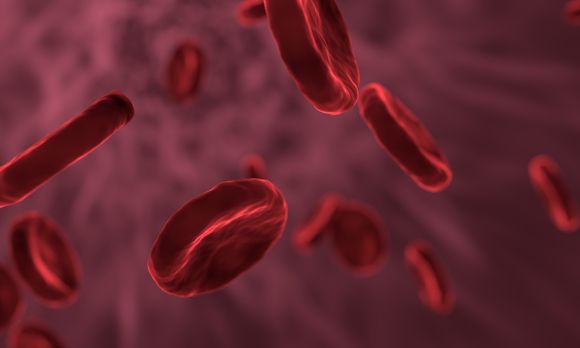
In sickle cell anemia, the mutation of hemoglobin is such that its structure has changed, and this gives erythrocytes unusual for them, but characteristic of the disease sickle form. The normal shape of erythrocytes is a biconcave disc.
This mutation is very common in Africa and other places around the world for which the widespread spread of malaria is characteristic, since the malaria parasite cannot enter and multiply in sickle shaped erythrocytes, and this provides malaria protection for sickle cell anaemia sufferers. Natural selection in this case leads to more people with the mutation, as they survive under the pressure of malaria.
What is Thalassemia
Thalassemia is a less common genetic disease, which leads to an insufficient amount of hemoglobin in red blood cells, as well as shorten their lifespan.
The condition causes anemia, a feeling of weakness and various other symptoms, depending on the type and severity of thalassemia. Thalassemia is two types, depending on which chain of protein hemoglobin is affected - alpha or beta. According to the severity of the disease, it is thalassemia minor, intermedia or major - mild, medium and severe form.
More about thalassemia read in the article Types of thalassemia, symptoms and treatment.
Herbs to Help Sickle Cell Disease Sufferers
As malaria is very common in Africa and therefore sickle cell anemia is, some of the herbs that are most studied and can be helpful also come from Africa.
Such medicinal plants are:
- Clove (Eugenia caryophyllata);
- Guinea pepper (Piper guineense);
- Grains of Paradise or Melegueta pepper (Aframomum melegueta);
- Sorghum bicolor also known as great millet, durra, jowari/jowar, or milo;
- Pterocarpus osun.
All of them, except for Grains of Paradise, are constituents of a drug used in sickle cell anemia called Niprisan. Phytocannabinoids (β-caryophylene) and vaniloids (eugenol, capsaicin and piperine) in cloves and Guinea pepper may explain some of the beneficial effects of Niprisan in sickle cell crisis - for example, pain relief. [ref.1]
Eugenol has an analgesic painkiller and antiseptic effect.
Capsaicin and piperine have almost no aroma, but they are very hot. It is interesting that the ability of these compounds to cause pain (i.e. burning sensation) makes them useful precisely for pain relief. Exposure to these substances reduces the sensitivity to pain and is used as a counteracting agent in the treatment of arthritis and other conditions with chronic pain.
It is known that over time a person becomes accustomed to hot food, and the body reacts to the pain of hot with the release of endorphins, which relieve pain and lead to pleasant feelings. This makes spicy food addictive.
Read more in the article .

Other herbs and fruits, that are used to improve the condition of sickle cell anemia sufferers, are:
- Root of the African tree Fagara zanthoxyloides;
- Unripe papaya fruits or leaves (Carica papaya);
- Garlic cloves (Allium sativum);
- Leaves of the African tropical tree Hymenocardia acida;
- Seeds of Pigeon peas (Cajanus cajan);
- Leaves or bark from the stem of the African tree Khaya senegalensis.
Vitamins and Minerals to Help Thalassemia Sufferers
Due to hemolytic anemia, increased nutritional needs, iron overload, diabetes and the use of chelators to remove excess iron, food deficiencies are common in thalassemia sufferers.
It is necessary to check the levels of calcium, vitamin D, vitamins E (alpha- and gamma-tocopherol) and C (plasma ascorbate), albumin, fasting glucose, fasting plasma zinc, serum copper and ceruloplasmin, serum selenium and serum folate once a year. [ref.2]
When taking multivitamin supplements, it is important that they are iron-free.
Patients with a mild form of thalassemia who do not receive a blood transfusion are recommended to take folic acid and a diet low in iron - avoid iron-fortified cereals and other products and excessive consumption of red meat. Drinking black tea during meals is recommended to reduce iron absorption from food.
For patients who get blood transfusions and are on chelation therapy, a low-iron diet is unnecessary and can impair quality of life.
Vitamin D intake is recommended for patients with 25-hydroxy vitamin D below 20 ng/dL.
In case of insufficient intake of calcium, it can also be obtained with supplements.
It is good to keep in mind that alcohol and cigarettes are dangerous for sufferers of thalassemia, since alcohol can lead to damage to liver tissue, and smoking cigarettes can lead to osteoporosis.

Sickle cell anemia and thalassaemia are genetic diseases affecting hemoglobin, but due to the large difference in severity of different forms of these diseases, they can allow normal life without the need for special therapy, but may also present with severe symptoms and complex treatment might be needed. In any case, appropriate dietary supplements or herbs may improve the quality of life of those affected, of course under medical supervision and with monitoring of relevant blood indicators.
Q&A
What is silent thalassaemia?
In silent thalassaemia there are no pronounced symptoms. One gene is missing or damaged, and the other 3 are normal. Blood tests are usually normal. Red blood cells may be smaller than normal. A parent with silent thalassaemia may pass on the damaged gene to their child. This is confirmed by DNA tests. [ref.3]
Thalassemia and pregnancy - what should you know?
During pregnancy, the volume of blood in the mother's body increases, which can lead to anemia and difficulties for the heart. Heart activity, as well as the liver functions of a pregnant woman with thalassaemia should be closely monitored throughout pregnancy. Taking folic acid is mandatory, as for any other pregnant woman in the first three months, as well as in the period before pregnancy.
A doctor is consulted about the likelihood that the baby has inherited the disease, especially if the partner also turns out to be carrying a defective gene.

How is sickle cell anemia inherited?
In order to develop sickle cell anemia, the child must have inherited one allel of the gene for the disease from both parents. Parents may not suffer from the disease, but be only carriers, that is, have only 1 defective allele of the gene and lack of symptoms. To inherit the disease, it is sufficient for one parent to suffer from sickle cell anemia (to have 2 allele defects) and for the other not to suffer but to carry the defective gene.
If both parents are carriers of the mutation, the risk of disease in their children is as follows [ref.4]:
- Probability that 1 in 4 every child they have will not inherit genes of the disease and have no sickle cell anemia or be able to transmit it;
- Probability 1 in 2 each child they have will simply inherit a copy of the sickle cell gene from 1 parent and be a carrier;
- Probability 1 in 4 each child they have will inherit copies of the disease gene from both parents and be born with sickle cell anemia.